Research Concept
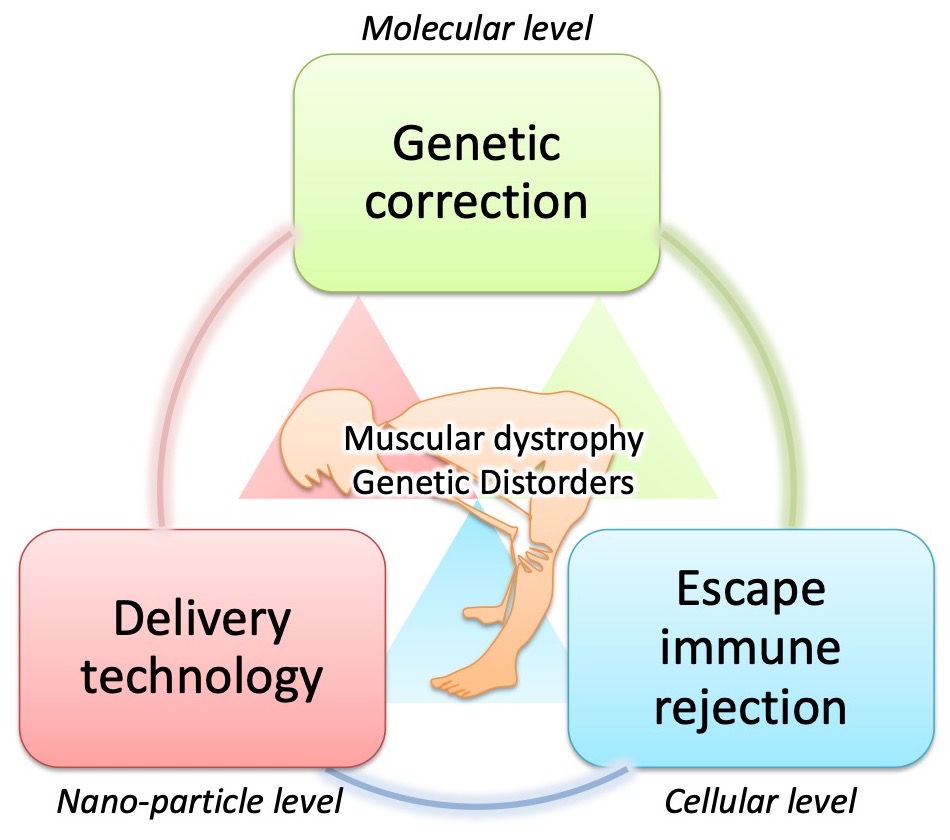 In order to realize gene therapy to repair gene mutations that are the root cause of intractable diseases, it is necessary to take various strategies according to the type of mutation or disease.
In our laboratory, we challenge these issues by applying gene delivery technology using viral and non-viral vectors, and genome editing technology such as CRISPR-Cas9. Here are some of the research projects conducted in the Hotta lab.
In order to realize gene therapy to repair gene mutations that are the root cause of intractable diseases, it is necessary to take various strategies according to the type of mutation or disease.
In our laboratory, we challenge these issues by applying gene delivery technology using viral and non-viral vectors, and genome editing technology such as CRISPR-Cas9. Here are some of the research projects conducted in the Hotta lab.
CRISPR-Cas9 Genome Editing
CRISPR-Cas is an adaptive immune system found in bacteria and archaea that helps defend against foreign nucleic acids that may inflict severe damage to the cell. Since there are many different types of Cas proteins, CRISPR-Cas systems are divided into 2 main classes based on the Cas signature genes and further divided into 6 types (I-VI). The Class 1 CRISPR comprises multiple effector complexes, while Class 2 contains a single protein effector. In 1987, Dr. Yoshizumi Ishino first discovered the crRNA sequence of Class 1 CRISPR in Escherichia coli. After 25 years, Drs. Emmanuelle Charpentier and Jennifer Doudna re-discover the Class 2 CRISPR as a programmable genome editing tool. In 2020, they won the Nobel Prize in Chemistry for the discovery of the Class 2 CRISPR-Cas9 system, which is easier to use in various research as it is composed of only a single protein factor, Cas9.
In the right figure, gRNA (guide RNA, purple) binds to target DNA (red and orange) to find the target sequence on the genome, Cas9 protein (green) DNA cleavage domain to induce DNA cleavage at the target site. By freely engineering gRNA responsible for DNA binding, DNA damage can be induced at any position on the human genome, to induce deletions or insertion.
NanoMEDIC: A transient delivery system packaged with Cas9 and gRNA in an extracellular vesicle
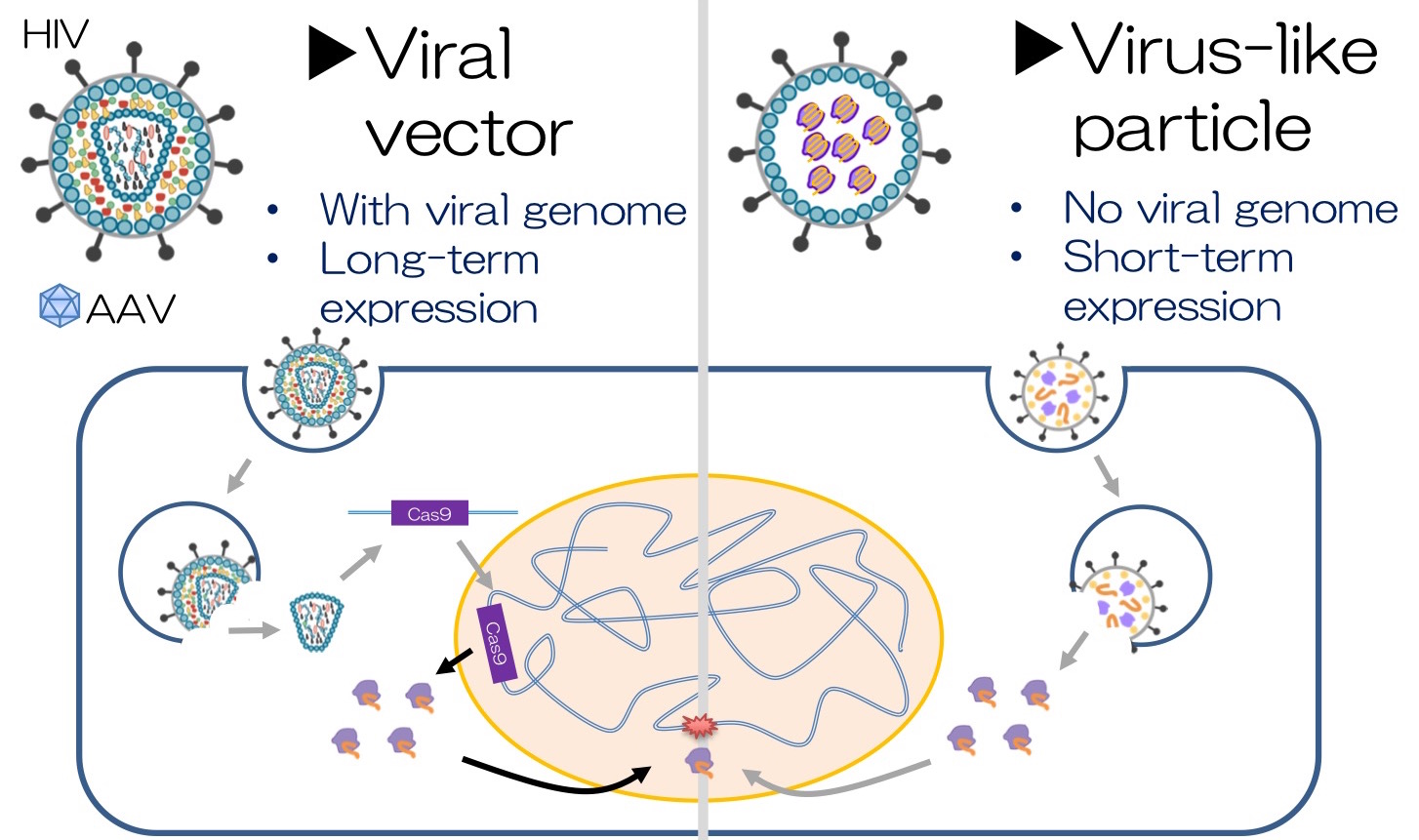 CRISPR-Cas9 system is a potential therapeutic approach to correct genetic mutations in diseases such as Duchenne muscular dystrophy (DMD). The most common methods to deliver CRISPR in the gene therapy field are Adeno-associated virus (AAV) vectors. However, overexpression from those vectors can persist for as long as several years. If Cas9 is overexpressed, it may cleave and edit non-targeted genes. To reduce these outcomes, a new method is essential to efficiently deliver CRISPR-Cas9.
CRISPR-Cas9 system is a potential therapeutic approach to correct genetic mutations in diseases such as Duchenne muscular dystrophy (DMD). The most common methods to deliver CRISPR in the gene therapy field are Adeno-associated virus (AAV) vectors. However, overexpression from those vectors can persist for as long as several years. If Cas9 is overexpressed, it may cleave and edit non-targeted genes. To reduce these outcomes, a new method is essential to efficiently deliver CRISPR-Cas9.
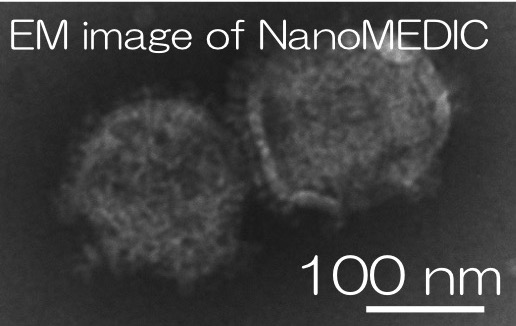
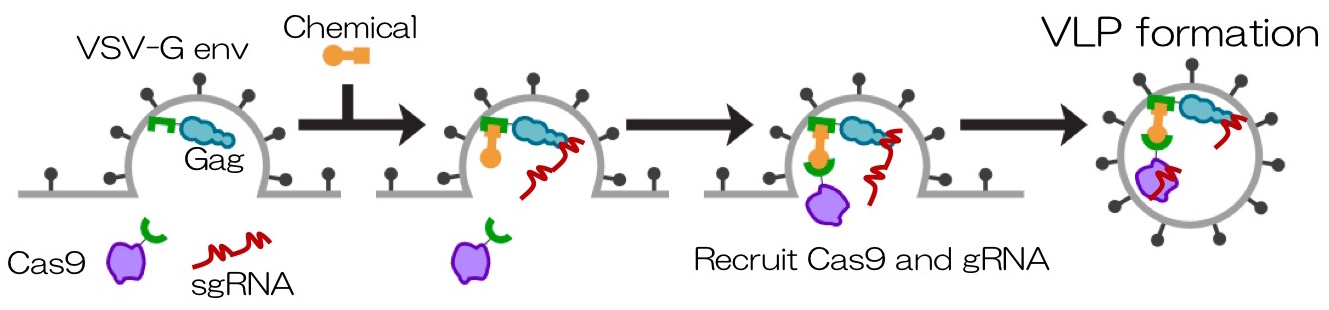 We developed a transient delivery system called NanoMEDIC which stands for Nano Membrane-derived Extracellular vesicles for Delivery of macromolecular Cargo. It is a virus-like particle (VLP), which is an ‘empty’ virus without its genome and has a transient expression which is an advantage to prevent prolonged expression of a transgene. We designed the VLP that can package Cas9/gRNA and efficiently edit targeted genes. First, we looked into a chemically inducible dimerization system FKBP12/FRB, where the rapamycin analogue drug mediates the binding of FRB and FKBP12. By fusing the FRB with Cas9 protein and the FKBP12 with HIV Gag protein, when rapamycin analogue is added, the Cas9/FRB is drawn to the Gag particles that form the core of VLPs. The HIV packaging signal was linked to gRNA so that it binds to Gag and enters inside of VLPs. Within VLP, sgRNA is excised by self-cleaving ribozymes to form a complex with Cas9.
We developed a transient delivery system called NanoMEDIC which stands for Nano Membrane-derived Extracellular vesicles for Delivery of macromolecular Cargo. It is a virus-like particle (VLP), which is an ‘empty’ virus without its genome and has a transient expression which is an advantage to prevent prolonged expression of a transgene. We designed the VLP that can package Cas9/gRNA and efficiently edit targeted genes. First, we looked into a chemically inducible dimerization system FKBP12/FRB, where the rapamycin analogue drug mediates the binding of FRB and FKBP12. By fusing the FRB with Cas9 protein and the FKBP12 with HIV Gag protein, when rapamycin analogue is added, the Cas9/FRB is drawn to the Gag particles that form the core of VLPs. The HIV packaging signal was linked to gRNA so that it binds to Gag and enters inside of VLPs. Within VLP, sgRNA is excised by self-cleaving ribozymes to form a complex with Cas9.
 We applied the NanoMEDIC delivery of Cas9/gRNA to various cells, such as skeletal muscles and neurons derived from patient iPSCs. In general, delivering a transgene in non-dividing cells is difficult, but we were able to deliver Cas9/gRNA and efficiently edit genes in iPS cell derived neurons.
We applied the NanoMEDIC delivery of Cas9/gRNA to various cells, such as skeletal muscles and neurons derived from patient iPSCs. In general, delivering a transgene in non-dividing cells is difficult, but we were able to deliver Cas9/gRNA and efficiently edit genes in iPS cell derived neurons.
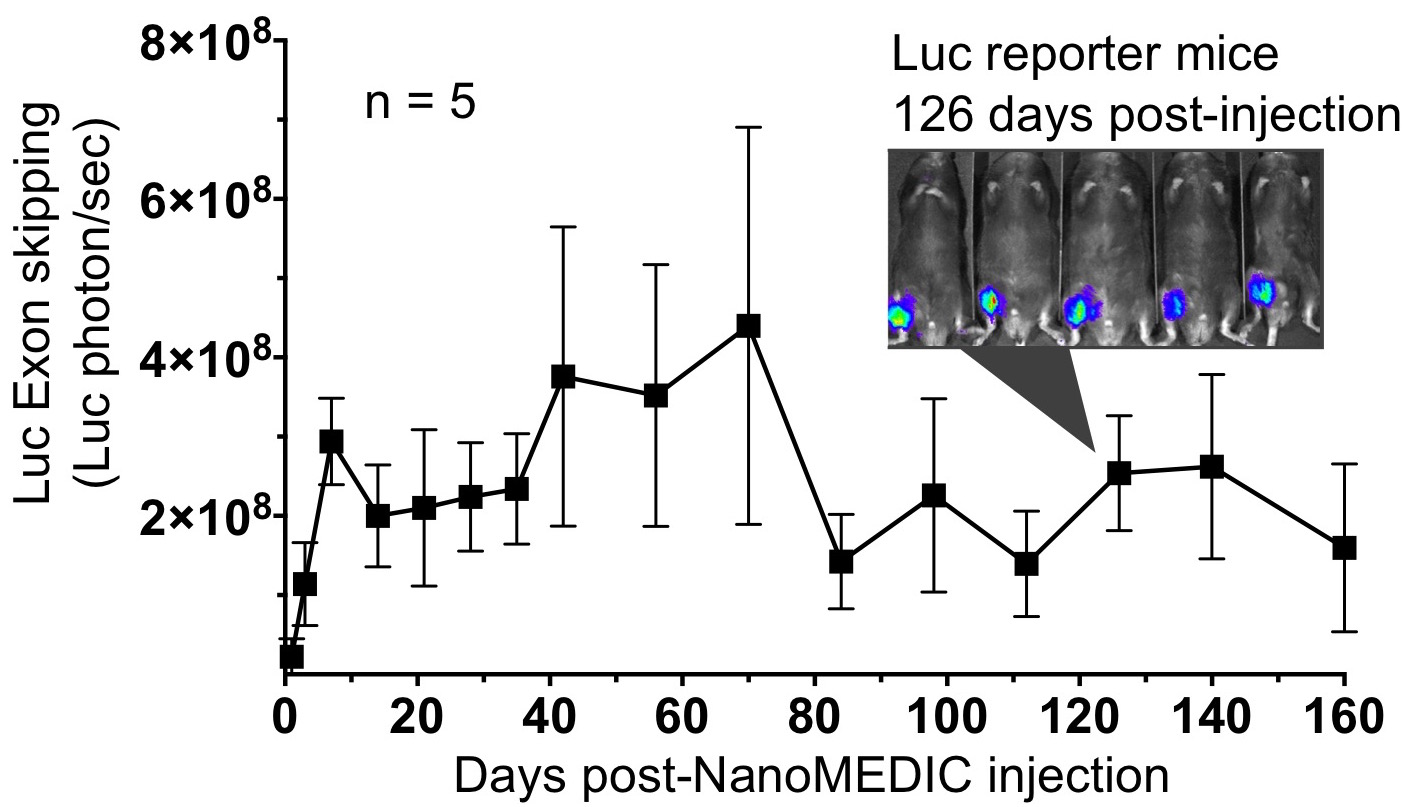 In addition, to determine the delivery efficiency in vivo, we used NanoMEDIC to induce exon skipping of the dystrophin gene in luciferase reporter mice. This reporter mice would emit firefly luciferase only when exon skipping is successful. We administrated the NanoMEDIC in the posterior compartment of the leg and a single treatment was sufficient to maintain the luciferase signal last for more than 160 days. Exon skipping was also successful in DMD model mice (mdx) as well.
In addition, to determine the delivery efficiency in vivo, we used NanoMEDIC to induce exon skipping of the dystrophin gene in luciferase reporter mice. This reporter mice would emit firefly luciferase only when exon skipping is successful. We administrated the NanoMEDIC in the posterior compartment of the leg and a single treatment was sufficient to maintain the luciferase signal last for more than 160 days. Exon skipping was also successful in DMD model mice (mdx) as well.
In summary, NanoMEDIC is an efficient method to deliver Cas9 protein and gRNA while reducing the risk of off-target mutagenesis compared to AAV or other virus vectors. In the future, we want to use the unique features of NanoMEDIC to treat DMD patients and other targets to advance the potential of genome editing therapy.
This project was published in Nature Communications (Mar 13, 2020)
Gee P, Lung MSY, Okuzaki Y, Sasakawa N, Iguchi T, Makita Y, Hozumi H, Miura Y, Yang LF, Iwasaki M, Wang XH, Waller MA, Shirai N, Abe YO, Fujita Y, Watanabe K, Kagita A, Iwabuchi KA, Yasuda M, Xu H, Noda T, Komano J, Sakurai H, Inukai N, and .Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping.
Nature Communications, 2020 Mar 13;. doi: 10.1038/s41467-020-14957-y.
[PubMed link] [Journal link]
Development of CRISPR-Cas3 System and Its Potential Application for DMD therapy
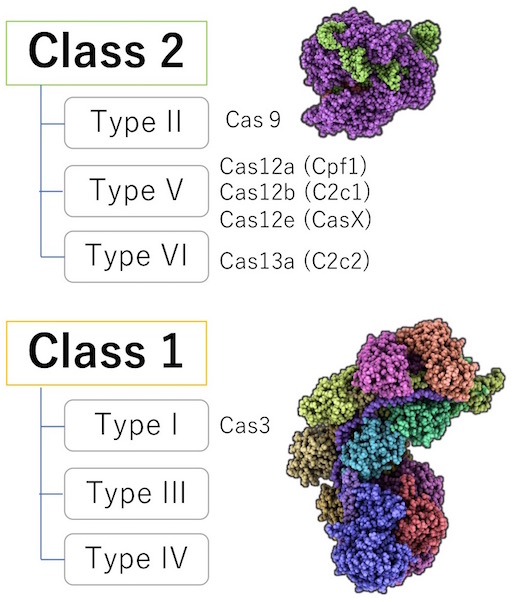 Our lab focuses on Class 1 for a few reasons. First, about 90% of the bacteria and archaea utilize the Class 1 system. It is thought that Class 1, unlike Class 2, has better DNA recognition and cleavage system that can differentiate foreign DNA from their own, also known as, the “self- versus non-self-recognition.” In addition, Cas9 is known to have off-target mutation risk and have complications with intellectual property. However, Class 1, unlike Class 2, does not have much evidence of whether a genetic modification in humans is possible. Hence, we took the chance and investigated this unexplored area.
Our lab focuses on Class 1 for a few reasons. First, about 90% of the bacteria and archaea utilize the Class 1 system. It is thought that Class 1, unlike Class 2, has better DNA recognition and cleavage system that can differentiate foreign DNA from their own, also known as, the “self- versus non-self-recognition.” In addition, Cas9 is known to have off-target mutation risk and have complications with intellectual property. However, Class 1, unlike Class 2, does not have much evidence of whether a genetic modification in humans is possible. Hence, we took the chance and investigated this unexplored area.
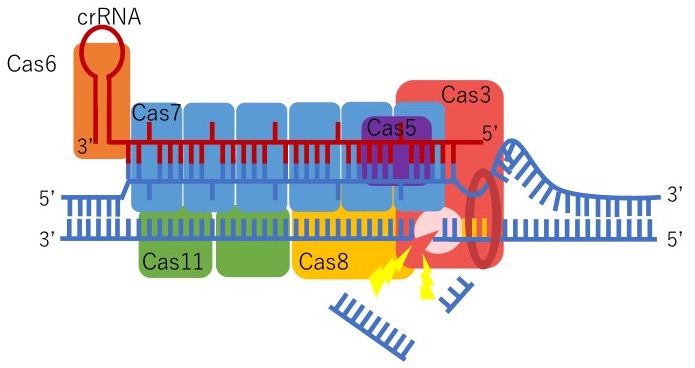 We were the first to discover that CRISPR-Cas3 can genetically edit human iPSCs by collaborating with Drs. Tomoji Mashimo (from Tokyo University) and Junji Takeda (from Osaka University). Type I-E CRISPR is derived from E.coli and forms a Cascade, or complex, with five proteins: Cas5, Cas6, Cas7, Cas8/Cse1, and Cas11/Cse2. This Cascade activates Cas3 which can degrade a remarkable distance of the targeted upstream DNA region. Also, genome-wide sequencing data indicate that Cas3 has fewer off-target mutations compared to Cas9. We believe this is because of two reasons: the length of crRNA and the mechanism to activate Cas3. The length of the crRNA, a guide for CRISPR-Cas3 to recognize foreign DNA, for Cas3 (27 base pairs) is longer than Cas9 (20 base pairs). This means that Cas3 can precisely identify the DNA sequences more than Cas9, hence, the reduced risk of off-target mutations. In addition, to activate Cas3, the Cascade initially needs to be formed with five proteins which include multiple steps. If there is even the smallest mistake in this process, Cas3 will not function.
We were the first to discover that CRISPR-Cas3 can genetically edit human iPSCs by collaborating with Drs. Tomoji Mashimo (from Tokyo University) and Junji Takeda (from Osaka University). Type I-E CRISPR is derived from E.coli and forms a Cascade, or complex, with five proteins: Cas5, Cas6, Cas7, Cas8/Cse1, and Cas11/Cse2. This Cascade activates Cas3 which can degrade a remarkable distance of the targeted upstream DNA region. Also, genome-wide sequencing data indicate that Cas3 has fewer off-target mutations compared to Cas9. We believe this is because of two reasons: the length of crRNA and the mechanism to activate Cas3. The length of the crRNA, a guide for CRISPR-Cas3 to recognize foreign DNA, for Cas3 (27 base pairs) is longer than Cas9 (20 base pairs). This means that Cas3 can precisely identify the DNA sequences more than Cas9, hence, the reduced risk of off-target mutations. In addition, to activate Cas3, the Cascade initially needs to be formed with five proteins which include multiple steps. If there is even the smallest mistake in this process, Cas3 will not function.
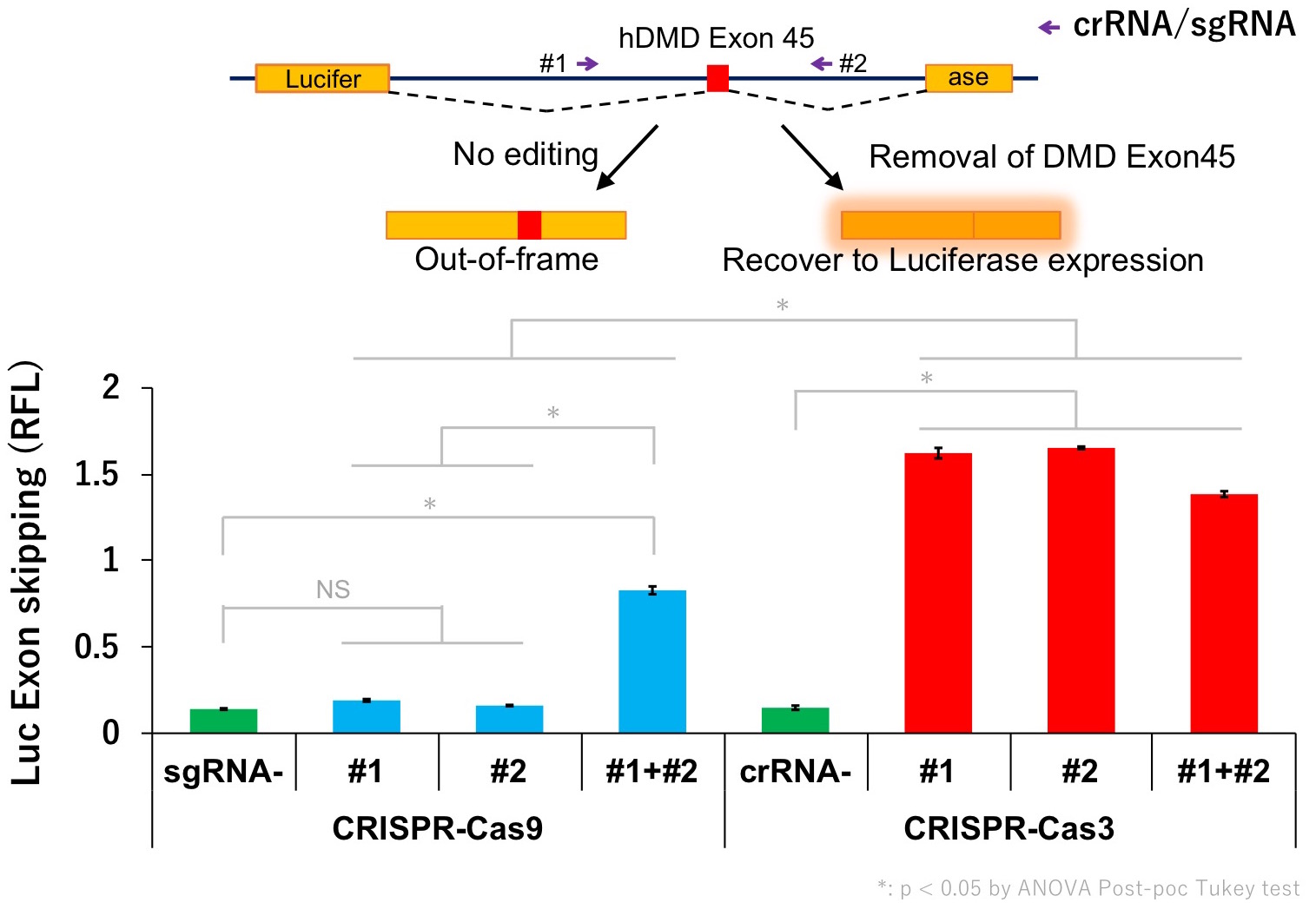 Currently, the mechanisms of the Cas3 system are not well understood yet and further research is necessary. Our next goal is to develop better gene therapy for DMD and further apply the unique characteristics of Cas3 in various fields.
Currently, the mechanisms of the Cas3 system are not well understood yet and further research is necessary. Our next goal is to develop better gene therapy for DMD and further apply the unique characteristics of Cas3 in various fields.
This project was published in Nature Communications (Dec 6, 2019)
 Morisaka H*, Yoshimi K*, Okuzaki Y*, Gee P, Kunihiro Y, Sonpho E, Xu H, Sasakawa N, Naito Y, Nakada S, Yamamoto T, Sano S, §, Takeda J§, and Mashimo T§. (*: Co-1st author, §: Co-correspondence)
Morisaka H*, Yoshimi K*, Okuzaki Y*, Gee P, Kunihiro Y, Sonpho E, Xu H, Sasakawa N, Naito Y, Nakada S, Yamamoto T, Sano S, §, Takeda J§, and Mashimo T§. (*: Co-1st author, §: Co-correspondence)CRISPR-Cas3 induces broad and unidirectional genome editing in human cells.
Nature Communications, 2019 Dec 6;10(1):5302. doi: 10.1038/s41467-019-13226-x.
[PubMed link] [Journal link]
HLA Genome Editing to Improve Immunocompatibility of iPS cells
When iPSCs from a donor are applied to a different patient, it may cause immune rejection due to a mismatch in Human Leukocyte Antigen (HLA or MHC) proteins which are located on the cell surface. To reduce such rejection, generating iPSCs from HLA homozygous donors would increase the HLA compatibility. Currently, CiRA (Center for iPS Cell Research and Application) at Kyoto University is producing HLA homozygous iPSCs that can be used especially among Japanese patients. In order to cover more than 90% of Japan's population, an estimate of 140 HLA-A, -B, and -DR homozygous iPSC lines need to be made. However, it is rare to find such donors and the generation of 140 iPSC lines will cost significantly. Another option to increase the immunocompatibility of iPSC is to knock out the B2M gene. B2M protein is involved in presenting HLA protein on the cell surface. Hence, silencing the B2M gene will lead to depletion of cell surface HLA proteins and will avoid being attacked by killer T Cells (cytotoxic CD8+). However, the absence of HLA will alert natural killer (NK) cells and such NK cells may attack the cells. Furthermore, the loss of all HLA proteins (on the cell) may compromise the immune system which may struggle to fight off tumours or infection.
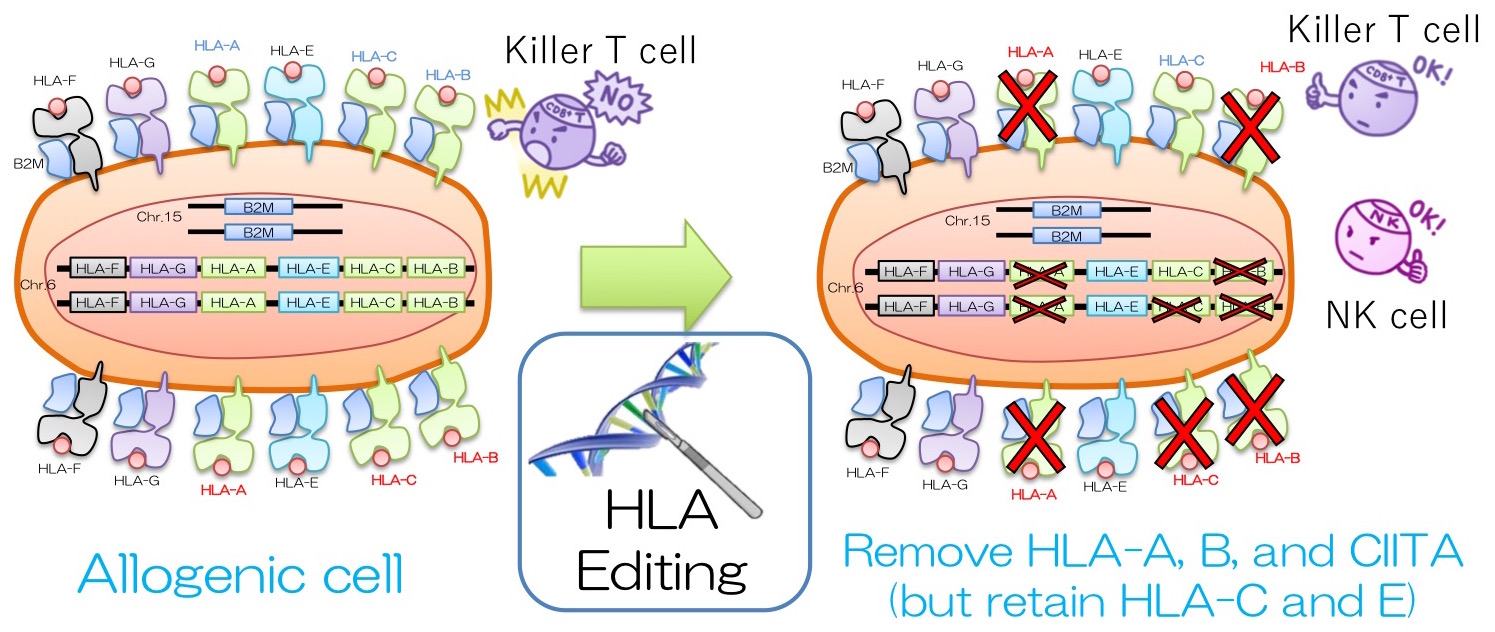 We created “HLA-C-retained iPSCs,” which maintain HLA-C expression (and other non-classical HLAs, such as HLA-E, -F, and -G), since they are known to suppress NK cells. It was made by deleting the HLA-A, and -B genes biallelically with CRISPR-Cas 9. Using these edited cells, we differentiated into blood cells and mixed them with the HLA-mismatched human CD8+ T cells and confirmed that the survival rate was high compared to the non-edited cells. We conducted another experiment and mixed the edited and differentiated cells with human NK cells. The data showed that the HLA-C-retained iPSCs were better at avoiding cytotoxicity than the B2M knockout (HLA-null) cells. Furthermore, we modified the HLA-C-retained iPSCs by deleting the CIITA gene, which is a transcription regulator for HLA Class 2 genes, to avoid activation of CD4+ helper T cells. Then we mixed the modified cells with the iPSC-derived blood cells and detected that there were no activities in CD8+ T cells, CD4+ T cells, and NK cells.
We created “HLA-C-retained iPSCs,” which maintain HLA-C expression (and other non-classical HLAs, such as HLA-E, -F, and -G), since they are known to suppress NK cells. It was made by deleting the HLA-A, and -B genes biallelically with CRISPR-Cas 9. Using these edited cells, we differentiated into blood cells and mixed them with the HLA-mismatched human CD8+ T cells and confirmed that the survival rate was high compared to the non-edited cells. We conducted another experiment and mixed the edited and differentiated cells with human NK cells. The data showed that the HLA-C-retained iPSCs were better at avoiding cytotoxicity than the B2M knockout (HLA-null) cells. Furthermore, we modified the HLA-C-retained iPSCs by deleting the CIITA gene, which is a transcription regulator for HLA Class 2 genes, to avoid activation of CD4+ helper T cells. Then we mixed the modified cells with the iPSC-derived blood cells and detected that there were no activities in CD8+ T cells, CD4+ T cells, and NK cells.
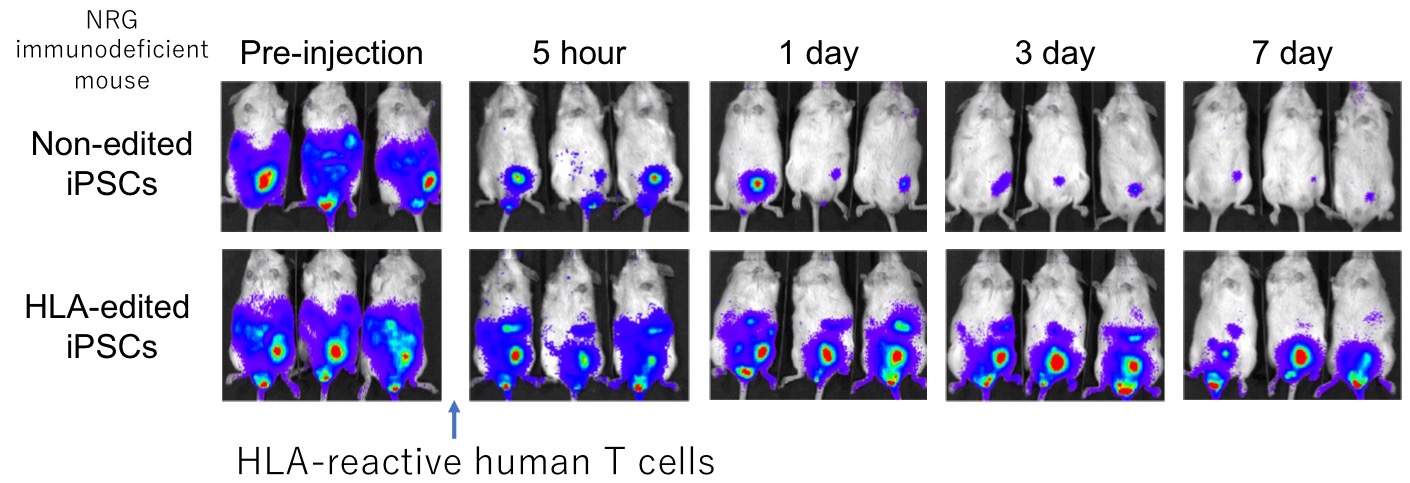
Moreover, we recreated the immune rejection in mice by differentiating the HLA-C-retained iPSCs into blood cells. Then we transplanted them into mice that lack immune cells but treated them with HLA reactive human T cells. As in the image, we determined that the survival rate was better in genome edited cells since it evaded cytotoxic activities from T cells.
By utilizing genome editing, we propelled a step forward in immunocompatibility with iPSCs. If we choose the appropriate HLA-C type with “HLA-C-retained iPSCs + deficient CIITA,” it is estimated that 6 iPSC lines will cover more than 90% of Japan’s population and 12 stable cell lines will cover more than 90% of every ethnic group we assessed. If a single iPS cell line can be used for a greater number of patients, it will lower the manufacturing cost. In addition, the avoidance of immune rejection would enhance the therapeutic effect of iPSCs with a prolonged survival rate.
This project was published in Cell Stem Cell (Mar 7, 2019)
 Xu H*, Wang B*, Ono M, Kagita A, Fujii K, Sasakawa N, Ueda T, Gee P, Nishikawa M, Nomura M, Kitaoka F, Takahashi T, Okita K, Yoshida Y, Kaneko S§, and §. (*: Co-1st author, §: Co-correspondance)
Xu H*, Wang B*, Ono M, Kagita A, Fujii K, Sasakawa N, Ueda T, Gee P, Nishikawa M, Nomura M, Kitaoka F, Takahashi T, Okita K, Yoshida Y, Kaneko S§, and §. (*: Co-1st author, §: Co-correspondance)Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility.
Cell Stem Cell, 2019 Mar 7;. doi: 10.1016/j.stem.2019.02.005.
[PubMed link] [Journal link]
Genome Editing Therapy for Muscular Dystrophy
Duchenne Muscular Dystrophy (DMD) is a severe muscle degeneration disease caused by the loss-of-function mutations in the Dystrophin gene on the X chromosome. Dystrophin is one of the biggest protein-coding genes consisting of 2 million bases. Exon skipping to modulate mRNA splicing patterns using antisense oligonucleotide is a promising approach currently tested in clinical trials, however, the effect of antisense oligos is transient. We aim to restore the mutated dystrophin protein using the programmable nucleases, such as TALEN or CRISPR-Cas9 system, in the patient-derived induced pluripotent stem (iPS) cells as a model.
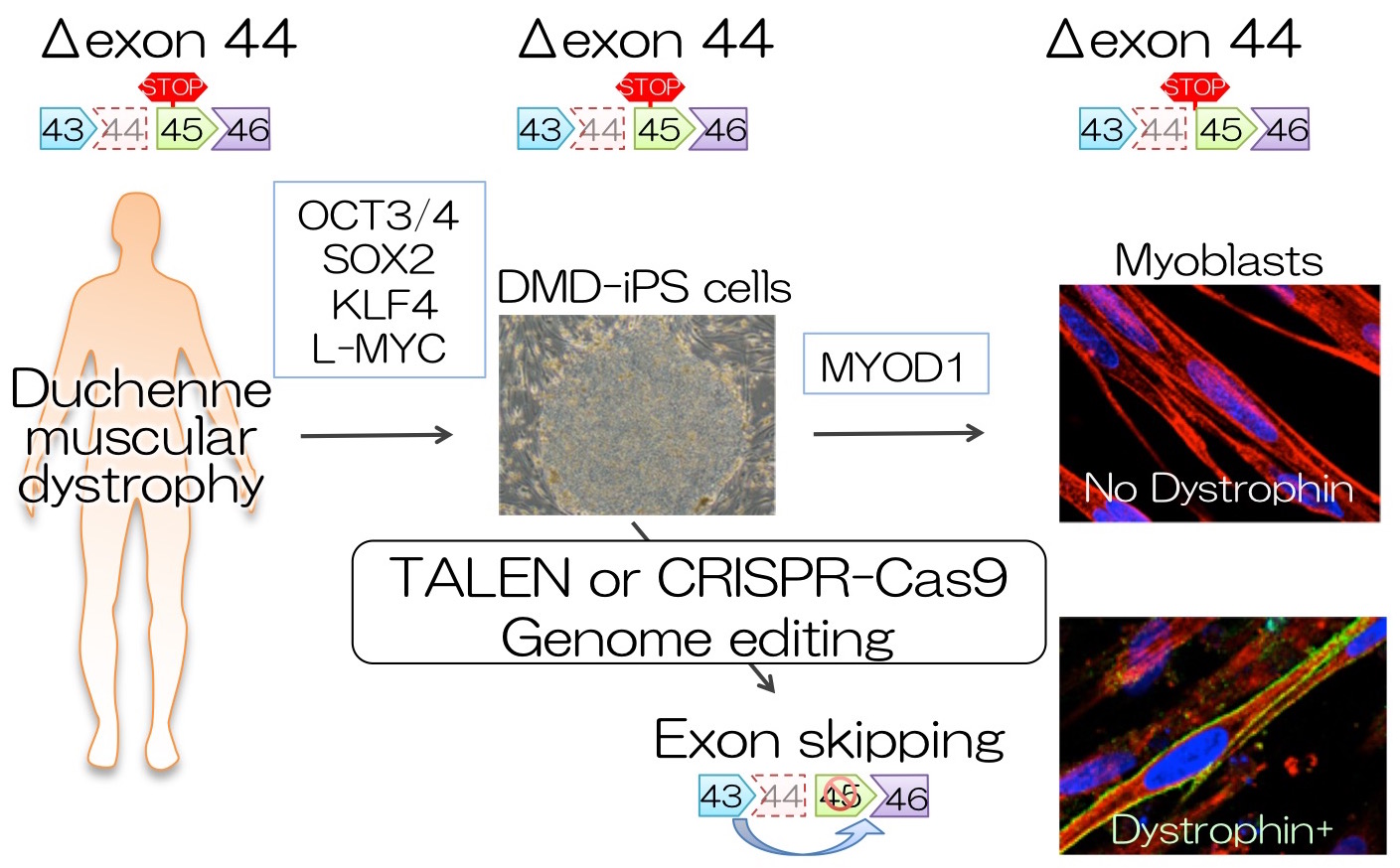
We first searched the human genome sequence in silico and extracted only a short DNA sequence of 10-16 bases in length on the human genome to create the iGEATs database. By using the iGEATs database, TALEN and CRISPR recognition sequences can be found in regions that can specifically target only one place on the human genome. Next, iPS cells were generated from a patient with muscular dystrophy and showed that the mutation of the dystrophin gene can be repaired using three strategies. Furthermore, when we examined the repair efficiency with TALEN and CRISPR, we found that either is effective. Next, we analyzed the karyotype of the chromosome, copy number variation of genomic DNA, and base mutation of the protein-coding regions in the iPS cells that were repaired, but no significant mutation was found. This result shows that genome editing can be performed with few side effects in iPS cells if TALEN and CRISPR are correctly designed and targeted. Finally, when the original and repaired iPS cells were differentiated into skeletal muscle cells, no expression of dystrophin protein was observed in the original iPS cells, whereas the dystrophin protein was detected in iPS cells after gene repair to a similar level as a healthy person. This result is expected to be a novel method of gene therapy for Duchenne muscular dystrophy.
This project was published in Stem Cell Reports. (Nov 26, 2014)
 Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T, Tanaka M, Amano N, Watanabe A, Sakurai H, Yamamoto T, Yamanaka S, and
Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T, Tanaka M, Amano N, Watanabe A, Sakurai H, Yamamoto T, Yamanaka S, and
Precise correction of the Dystrophin gene in Duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9.
Stem Cell Reports, 2015; Vol.4 (1): p143-154. [PubMed link] [Journal link]
Derivation of human iPS cells with EOS pluripotency reporter
iPS cells can be generated from adult somatic cells by introducing a cocktail of transcriptional factors, and have an enormous potential for future stem cell therapy. However, induction efficiency of iPS cells is still low (~0.02%), and the heterogeneous nature of reprogramming makes it difficult to control the quality of the iPS cell lines and their differentiation potentials.
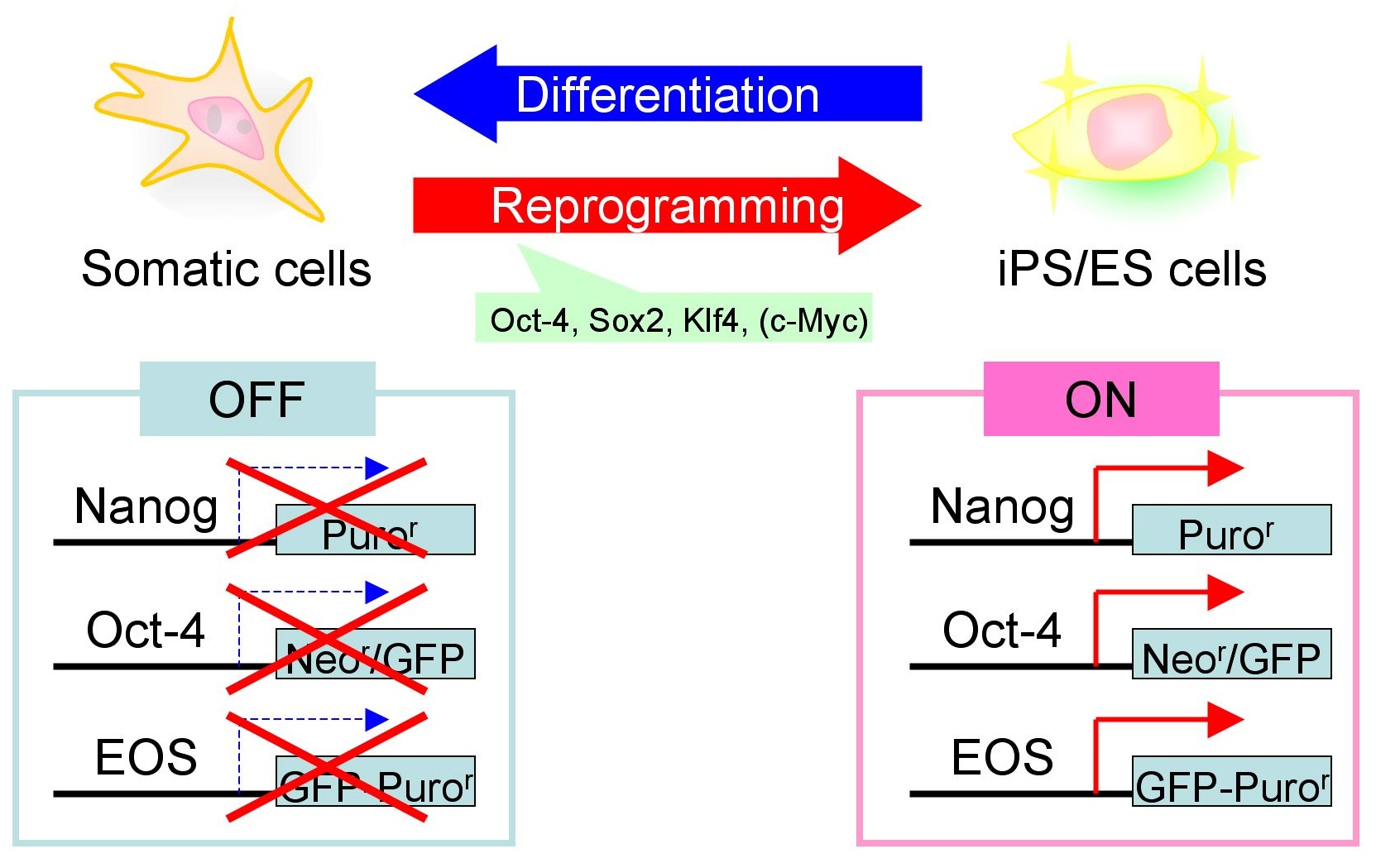
When I was in the Ellis lab at Toronto, I have developed a novel selection system for human iPS cells by using a lentiviral vector that specifically expresses GFP (green fluorescence protein) gene in pluripotent stem cells. By utilizing Early Transposon (ETn) promoter combined with Oct-4 (= Pou5f1) core enhancer elements, resultant EOS lentiviral vector can express GFP specifically in pluripotent stem cells but extinguished after differentiation.
When the EOS vector was introduced into human fibroblasts, GFP was not activated as expected. However, after induction of iPS cell reprogramming by a forced expression of the Yamanaka factors (OCT-4, SOX2, KLF4, C-MYC), GFP expression was activated on emerged iPS cell colonies. By selecting for puromycin resistance gene which is under the control of the EOS cassette, only iPS cells can be grown and enriched the percentage of hESC marker (i.e. TRA-1-81) positive iPSC population up to 70%. We also succeeded to isolate iPS cell lines from Rett Syndrome patient who has neurodevelopmental disorder due to a missense mutation in the MeCP2 gene. The EOS vector is not only useful for selecting iPS cells, but also has the potential to utilize for optimizing novel induction methods and for screening small molecules to enhance reprogramming.
This project was published in Nature Methods. (April 26, 2009)
 , Cheung, AY, Farra, N, Vijayaragavan, K, Seguin, CA, Draper, JS, Pasceri, P, Maksakova, IA, Mager, DL, Rossant, J, Bhatia, M, Ellis, J.
, Cheung, AY, Farra, N, Vijayaragavan, K, Seguin, CA, Draper, JS, Pasceri, P, Maksakova, IA, Mager, DL, Rossant, J, Bhatia, M, Ellis, J.Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency.
Nature Methods, 2009; Vol.6 (5): p370-376. [PubMed link] [Journal link]